|
MC-TB 2010 Vol 2, BTHM2010-c1
FrameShift_Viewer:分析diploid DNA中single DNA deletion軟體
連掌豪、李御賢*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:李御賢[ bojack@mail.mcu.edu.tw ]
收稿:2010-4-8 接受:2010-4-9
中文摘要
本軟體嘗試利用human genome DNA,利用同一組primers以PCR取得diploid DNA 中序列,但是在不擷取haploid DNA 片段再定序的情況下,因為分別來自父母的diploid DNA 不會具有完全相同的序列。在diploid DNA 長度不同情況下PCR product定序會有訊號重疊的情況產生。序列判讀上很難將diploid DNA序列區分。 本軟體以MATLAB撰寫,能夠自動連接各種生物資料庫(如:NCBI)取得haploid DNA 序列,利用電腦快速計算的特性,讓軟體載入ABI 定序檔 [1],並自動化分析、 模擬演算,檢測出diploid DNA 實際序列。
緒 論
DNA polymorphism 在近年來一直被廣泛探討,認為與個體的phenotype 息息相關。因此在研究這些課題上,序列分析技術如PCR、real-time Q-PCR、multiplex ligation-dependent probe amplification (MLPA) 、array comparative genomic hybridization (array-CGH)、fluorescence in situ hybridization (FISH)不斷推陳出新, 其中以PCR最快速、低廉 [2]。我們發現當single nucleotide polymorphism(SNP)
若為base insertion or base deletion 只在任一haploid DNA 之一時,PCR products 定序時會發現訊號值兩組重疊,原因是兩條haploid DNA 序列其中一條shift。有了此項概念後,我們將這套規則以MATLAB撰寫成圖形化界面的軟體,目前已能順利完成human genome DNA在分析diploid DNA中single DNA deletion的自動化判斷分析。未來預計能擴展出two type HPV DNA mix自動化判斷分析系統,以及microsatellite repeat number 自動化判斷分析系統。
材料與方法
與長庚醫院合作取得human genome DNA 以及定序ABI data,經過SNP 篩選、primers設計、PCR、PCR product 定序後得到定序ABI data。(1)首先將ABI file 載入MATLAB 撰寫的圖形化介面軟體FrameShift_Viewer (2)程式再依據mix sequences會有兩個明顯較大的peaks,自動化判斷single sequence region (3)接下來軟體向NCBI Entrez自動取得該區域haploid DNA sequence (4)最後軟體會自動模擬演算以找出其中一條DNA deletion 的組合,並且mix sequences會完全符合軟體載入的ABI data。
結 論
目前對於mix sequences訊號值判定,同一點重疊率高的peaks,軟體的自動判斷性相當優良,並能得到正確的結果,但是對於重疊率較低的組合,未來將透過強化數學模組的方式,讓軟體能自動判讀複雜性高的data。
參考文獻
[1] http://www3.appliedbiosystems.com/AB_Home/index.htm.
[2] Gödde R, Akkad DA, Arning L, Dekomien G, Herchenbach J, Kunstmann E, Meins M, Wieczorek S, Epplen JT, Hoffjan S, 2006, Electrophoresis of DNA in human genetic diagnostics - state-of-the-art, alternatives and future prospects. Electrophoresis 27(5-6):939-46.
MC-TB 2010 Vol 2, BTHM2010-c2
Purification of lipidated Survivin protein
饒峻宇、吳慧中*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:吳慧中[ joannawu@mail.mcu.edu.tw ]
收稿:2010-4-9 接受:2010-4-9
中文摘要
本實驗選用survivin 基因,被證實會在大多數的腫瘤細胞中過度表現以抑制細胞凋亡。利用基因重組技術將不同長度的信號胜肽送入大腸桿菌菌種中,經由SDS-PAGE 分析以及西方轉漬法確認脂質化蛋白質,並使用Ni-NTA 管柱,以imidazole 析出蛋白質。
緒 論
現代疫苗以重組蛋白或合成胜肽被認為是安全且較少副作用。然而,這些疫苗其免疫性通常很差,需要加入佐劑來提升免疫能力。因此,為了加強重組蛋白的效果,利用基因工程設計一段免疫抗原,經三種酵素分別作用後,以完成脂質化修飾。survivin 基因為凋亡抑制(IAP)家族的成員之一,能抑制caspase 活化導致負調控細胞凋亡。survivin 蛋白會高度表達於腫瘤細胞中,此一事實成為理想的治療目標癌症的癌細胞為目標,而使正常細胞正常存活。選擇survivin 基因將其基因重組後,送入大腸桿菌菌種表現。以SDS-PAGE 分析和西方轉漬法確認蛋白質。觀察經轉譯後修飾的脂質化蛋白質表現。
材料與方法
選擇survivin 基因,在其N 端加入不同長度的信號胜肽,轉殖至大腸桿菌菌種內(C41 & C43),以10% SDS-PAGE 分析其表現的情形並用西方轉漬法確認蛋白質是否有表現。確認後,以C43D1 為目標,利用其含有6 個histidine 的片段,使用Ni-NTA 管柱以不同濃度梯度之imidazole 將其析出並用10% SDS-PAGE 分析後,再以N 端定序確認蛋白質脂質化的情形。
結 論
目前以4 種不同長度的信號肽分別送入大腸桿菌菌種內表現其脂質化的情形,選擇以片段最短經西方轉漬法確認,有脂質化蛋白質作為此次實驗的目標。以Bradford 定量為200 ng 後送至台大實驗室做N 端定序,確認其N 端是否被blocking。
參考文獻
[1] Leng et al, Vaccine, 2009, 25;27(9):1400-9.
[2] Rezwan et al, Microbiology, 2007, 153:652–658.
[3] Tamm et al, Cancer Research, 1998, 58:5315-5320
MC-TB 2010 Vol 2, BTHM2010-c3
人類諾羅病毒#406株之VP1基因選殖
余彥苓、張猷忠*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:張猷忠[ d80106@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-4-9
中文摘要
諾羅病毒(norovirus)是造成急性腸胃炎的重要病因。為了可以快速檢測出是否曾受到諾羅病毒的感染,所以希望用諾羅病毒的主要殼蛋白當作抗原,來檢驗病患血清檢體,確認是否曾遭受諾羅病毒的感染。實驗材料來自長庚兒童醫院所提供疑似諾羅病毒感染病患的檢體,經設計專一性引子,選殖出部分諾羅病毒主要殼蛋白序列,希望轉殖入載體(pMAL-c2x),以便病毒主要殼蛋白的表現及純化。
緒 論
諾羅病毒(norovirus)近年來被發現是造成急性腸胃炎群體感染的主要病毒病因,屬於Caliciviridae 家族,基因組全長大約為7.6 kb,是正股單股RNA (positive-sense single-stranded RNA)的無封套病毒,由三個開放讀序框 (open reading frame,ORF) 所組成的。ORF1 可以分別轉譯成p48、NTPase、p22、VPg、3CLpro 和RdRp;ORF2 轉譯成主要的殼蛋白VP1;ORF3 轉譯成次要的殼蛋白VP2。與人類疾病相關的基因型有兩個,分別為GI和GII,GI的ORF1 中可以轉譯成p22 蛋白,GII則可以轉譯成p20 蛋白。諾羅病毒尚無法培養在細胞中,但可以利用重組蛋白來表現病
毒的基因。 諾羅病毒所導致的急性腸胃炎,在人體的潛伏期為 24~48 小時。感染者通常會
出現嘔吐、腹瀉、腹絞痛和噁心等病徵。除了幼兒、老年人和免疫功能不足者,只要能適當的補充流失的水分,給予支持性治療,症狀都能在數天內改善。諾羅病毒於台灣的流行尚未清楚,但復原者對它會有持續好幾個月的專一性免疫。不同基因型的諾羅病毒,可能在個體中產生重複感染甚至影響終生。為瞭解民眾傳染諾羅病毒的情形,故希望選殖表現諾羅病毒主要殼蛋白做為抗原,冀能以酵素免疫分析法(enzyme immunoassay, EIA)來進行調查。
材料與方法
實驗材料是長庚兒童醫院所提供,來自疑似被諾羅病毒感染的病人檢體#406。先將PCR 選殖出的諾羅病毒主要殼蛋白序列與質體pUC19 黏合後,轉殖到大腸桿菌勝任細胞JM109 株,再篩選出有轉殖成功的菌落,送去定序確認後,再將主要殼蛋白序列選殖入表現載體pMAL-c2,然後表現並純化病毒蛋白做為抗原。
結 論
目前已可成功以PCR增幅出病毒的主要殼蛋白,選殖工作仍在進行中。
參考文獻
[1] Wobus et al, J. Virol, 2006, 80:5104.
[2] Rohayem et al, J. Virol, 2005, 79:4977.
[3] Asanaka et al, PNAS, 2005, 102:10327.
[4] Parker et al, J. Virol, 2005, 79:7402.
MC-TB 2010 Vol 2, BTHM2010-c4
利用水稻生產基因工程葡萄糖轉化酶以加速葡萄糖的製造
簡洧娟、吳怡萱、陳建成、黃國旭、江志明*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:江志銘[ cmchiang@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-3-18
中文摘要
在植物澱粉儲存器官大量表現耐熱的細菌來源澱粉代謝酵素,是一個用來改良澱粉品質、營養成份及分解流程的重要策略。我們利用農桿菌轉殖法轉殖耐熱的葡萄糖轉化酶到台農67 號水稻品種,使能得到葡萄糖轉化酶的轉殖水稻種子,用以生產葡萄糖。高葡萄糖糖漿可被廣泛的應用在生質酒精的製程。因其可節省工業上生產葡萄糖糖漿所需的時間與花費。更進一步可應用於澱粉含量更多的農作物上,此舉將對台灣的農業與食品工業有所助益。
緒 論
每年台灣地區生產稻米所留下的稻梗總數可高達 一百三十一萬噸。這些稻梗廢棄物通常都是以焚燒的方式處理,雖然如此能夠抑制稻米疾病的傳播,但其過程中所產生的廢氣,卻會造成許多像是氣喘類的呼吸性疾病。我們自Rhizopus oryzae 純化出 耐熱的 glucoamylase 基因,利用農桿菌轉殖入水稻中大量表現,利用它能降解 澱粉非還原端的 α-1,4 linkages 之能力,水解澱粉釋放出葡萄糖。可進一步被利用來生產生質酒精,改善農業廢棄物對環境所造成的污染。
材料與方法
材料:未成熟TNG 67 種子於MS 培養基(2,4-dichlorophenoxyacetic acid 2mg/L) 上培養、Top 10 E.coli strain 勝任細胞、DB 3.1 E.coli strain 勝任細胞、pENTR vectors、pPZP200 binary vectors、ccdB 基因片段。.表現載體的構築:我們利用Gateway cloning system 構築表現載體,此系統利用
site-specific recombination 的原理,不僅擁有不需用限制酶、連接酶及可以定向插入載體的優勢,而且省時、表現佳、任何vector 都可使用,故應用的層面廣泛。首先將glucoamylas 與pENTR vector 接合,經過細菌轉殖篩選後,以Gateway cloning system 將glucoamylase cloning 入pPZP200 binary vector。接著利用農桿菌感染未成熟稻米TNG67 種子的癒傷組織,並以hygromycin 確認glucoamylase 在植物中的表現。
結 論
目前glucoamylase 表現載體已利用農桿菌轉殖入水稻癒傷組織並以hygromycin 篩選確認中。待以PCR 分析得到轉殖株後,接著用北方墨點法分析基因插入的考貝數( copy number);南方墨點法測其在植株各組織的表現量,篩選出表現強的植株。將應用於食品或生質酒精的製造,改善農業廢棄物所造成的環境污染問題。
參考文獻
[1] 徐子勤,北京:科學出版社;2007。
[2] 關麗英等,Journal of Capital Normal University。2002,23:2。
[3] Lin et al, BMC Biochemistry, 2007, 8:9.
MC-TB 2010 Vol 2, BTHM2010-c5
利用16S rDNA來辨識砧板表面微生物
粘佳琪、張猷忠*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:張猷忠[ d80106@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-4-9
中文摘要
為了解不同材質砧板表面上的菌相與砧板使用情形之關聯,所以針對不同材質砧板表面的微生物進行菌相調查。利用聚合酶連鎖反應分析16S rDNA序列,已可鑑定砧板細菌,唯需增加選殖定序樣本數,始能得到較完整的菌相。
緒 論
因為砧板幾乎是天天都會使用到的器具,可直接接觸到我們所吃的食物,若能了解砧板使用情形與菌相之關聯,當有助益於食品衛生工作。16S rDNA 是一種目前常用鑑定微生物的分析方法[1, 2],可取代傳統培養過程而直接進行分析,解決傳統無法清楚鑑定之菌種,及克服某些菌種無法或難以培養之缺點,而得到較為完整的菌相圖譜。可從Ribosomal Database Project (RDP)資料庫中取得保留區域的基因序列,依據保留區域設計primer,利用聚合酶鏈反應,將砧版上微生物特定的16S rDNA 序列片段拿去複製放大,經過篩選及定序,將基因序列分析出來,並與NCBI資料庫進行比對,比對完就可以很快鑑定出砧版上微生物的物種。
材料與方法
首先採樣砧板上菌種,利用colony PCR萃取菌種的genomic DNA,接著做聚合酶連鎖反應,將聚合酶連鎖反應產物跑完洋菜膠體電泳可以確定是否有夾出我們要的片段,將有夾出來的band 切下來進行純化,使用yT&A Cloning Vector Kit 進行接合作用,將接合作用後之產物轉殖入勝任細胞(JM109 high efficiency competent cell)內,由勝任細胞進行transformation,利用藍白篩進行選殖是否有殖入成功,抽取殖入成功的plasmid,加入限制酶切解檢查16S rRNA gene 是否有插入正確的
結 論
初步送了兩個轉殖株去定序,有一株是從塑膠砧版取出去定序,定序出來可以判定是大腸桿菌,之後會再送多一點去定序。抽取質體最後是利用colony PCR的方法才成功,成功的可能原因或是因為沒有經過洗鹽等步驟,採樣來的樣本菌數原本就不多,不會經過較多實驗過程而流失。
參考文獻
[1] Bottger, E. C. FEMS Microbiol. Lett. 1989, 65:171.
[2] Chen et al, FEMS Microbiol. Lett. 1989, 57:19.
MC-TB 2010 Vol 2, BTHM2010-c6
利用定序16S rDNA來辨識金黃色葡萄球菌
林沛禔、張猷忠*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:張猷忠[ d80106@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-4-9
中文摘要
金黃色葡萄球菌(Staphylococcus aureus)為最常引起感染傷口的葡萄球菌,因此對於傷口感染的檢體快速的鑑定是非常重要的。因為由於S. aureus 為陽性菌,細胞壁的構造與陰性菌不同,在抽取genomic DNA 時較為困難,因此實驗中利用了phenol/chloroform extraction、alkaline-SDS lysis 及guanidium thiocyanate lysis 抽取,所顯現出的結果只有guanidium thiocyanate lysis 的方法能有效抽
取genomic DNA,並進一步進行菌種鑑定。
緒 論
Staphylococcus aureus 是最常引起感染的葡萄球菌,由於S. aureus 容易感染皮膚上的傷口,引起膿腫(膿及組織壞死),所以醫療院所對S. aureus 尤其注意。因此在感染傷口的檢體上需能快速的鑑定出菌種。一般傳統鑑定細菌上,都是以生化測試的方式去分類細菌,然而這種方式需要繁
雜的生化測試且又並非完全正確。此外,則要找出可以穩定的抽取genomic DNA的方法。金黃色葡萄球菌為革蘭氏陽性菌,因此細胞壁的組成與陰性菌不同,在抽取genomic DNA 較為困難,我們比較幾種抽取方法,找出較為有效的萃取金黃色葡萄球菌genomic DNA 的方法。
材料與方法
利用phenol/chloroform extraction、alkaline-SDS lysis 及guanidium thiocyanate lysis 等方法抽取genomic DNA。之後16S rDNA 通用引子(universal primers)進行PCR,產物經選殖定序後,以BLAST 比對鑑定菌種。
結 論
萃取genomic DNA 的方法,利用了phenol extraction、alkaline-SDS lysis,皆無法成功萃取出genomic DNA。可能與細胞壁組成有關,在參考之前文獻後[1, 2],利用guanidium thiocyanate 方法萃取出genomic DNA 後,PCR 可成功放大16S rDNA 片段。
參考文獻
[1] Bottger, E. C. FEMS Microbiol. Lett. 1989, 65:171.
[2] Pitcherd et al, Letters in Applied Microbiology 1989, 8:151-156
MC-TB 2010 Vol 2, BTHM2010-c7
利用16S rDNA Gene定序快速鑑定市售乳酸菌飲料所含菌種
施采蒨、張猷忠*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:張猷忠[ d80106@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-4-9
中文摘要
本實驗嘗試檢測市售乳酸菌飲料中的菌種。在不使用市售genomic DNA純化套組情形下,比較phenol/chloroform extraction、alkaline-SDS lysis、alkaline lysis combined phenol extraction 及guanidium thiocyanate lysis 抽取方法後,顯示前面三種方法無法萃取出乳酸菌的genomic DNA,只有guanidium thiocyanate lysis 可以有效萃取出genomic DNA,並經16S rDNA-PCR 證實可用於乳酸菌的快速檢測。
緒 論
市售乳酸飲品多宣稱含有各式乳酸菌,為了確認各類產品所含菌種是否和標示相同,爰進行本實驗。進行實驗須先抽取所含菌種的genomic DNA,目前市面上雖有多種抽取genomic DNA試劑套組,但之前實驗曾發現並非所有細菌皆可順利抽取,這或和細菌胞壁多醣類組成及含量有關,也是為了方便沒有套組時抽取乳酸菌核酸參考,所以嘗試並比較phenol/chloroform extraction、alkaline-SDS lysis、 及guanidium thiocyanate lysis [1]抽取方法。細菌鑑定則利用16S rDNA-PCR [2],定序PCR產物並以BLAST分析。所有的細菌都有含有16S rRNA,所以16S rDNA
基因序列進行比較,就可以協助做細菌的鑑定及分群。
材料與方法
購買市售不同產牌含乳酸菌飲料,分離細菌並比較phenol/chloroform extraction、alkaline-SDS lysis、alkaline lysis combined phenol extraction 及guanidium thiocyanate lysis 抽取genomic DNA方法。隨後以16S rDNA通用引子(universal primers)進行PCR,產物經選殖定序後,以BLAST比對鑑定菌種。
結 論
目前共取兩種飲料菌種樣本,先直接萃取genomic DNA跑PCR,但並未成功。原因可能是產品出售前添加殺菌劑,使得樣品內含菌量少或無,萃取出的genomic DNA 量不夠;先畫在MRS agar 上培養的樣本,因非培養在厭氧狀態,可能會使一些厭氧菌無法生存增殖。至於萃取genomic DNA 的方法,試過了phenol extraction、alkaline-SDS lysis、alkaline combined phenol extraction,皆無法成功萃取出genomic DNA。目前認為可能與G(+)陽性菌胞壁組成有關,在參考之前文獻後[2],利用guanidium thiocyanate方法成功萃取出genomic DNA,亦可以PCR 放大16S rDNA片段。
參考文獻
[1] Pitcherd et al, Lett. in Appl. Microbiol. 1989, 8:151.
[2] Bottger EC, FEMS Microbiol. Lett. 1989, 65:171.
MC-TB 2010 Vol 2, BTHM2010-8
利用16S rDNA Gene定序鑑定沙門氏菌
曾鈺婷、張猷忠*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:張猷忠[ d80106@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-4-9
中文摘要
相較於傳統生化試驗鑑定細菌,利用16S rDNA sequencing的方式可以更有效率及低成本的鑑定細菌。在所有原核生物16S rDNA 的序列中,有保守區域及異變區域,其中異變區域,可用生物親源性的差異來鑑定菌種。實驗利用PCR放大沙門氏菌16S rDNA 片段,再挑選轉殖成功的菌株定序比對鑑定。透過實驗設計可找出穩定抽取出genomic DNA 的方法,結果顯示利用alkaline 及phenol 兩者的結合,可以有效的抽取出沙門氏菌的genomic DNA。
緒 論
沙門氏菌的感染引發急性腸胃炎,在台灣地區的夏天為常見的傳染性疾病。發病後初期症狀為發燒、噁心、嘔吐、腹脹或腹絞痛,接著會出現帶有血絲或是黏液的腹瀉,嚴重時腹瀉可能導致脫水,甚至引發敗血症。傳統沙氏桿菌鑑定方法繁複,利用16S rDNA-PCR sequencing的方式可以更有效率、提高靈敏度、省時且低成本的對菌種做鑑定[1]。所有的細菌都含有16S rDNA序列,其中可分成保守區以及易變區,在保守區域內都具有高度相似的序列,這可以用來設計通用引子(universal primers),再利用異變區的差異對細菌作序列上的比對,找出他們的親源性關係,來鑑定菌種[2]。此外,則要找出可以穩定的抽取genomic DNA的方法。沙門氏菌為革蘭氏陰性菌,不同種革蘭氏陰性菌胞壁所含多醣類不一定相同,可能會影響隨後進行實驗的酵素功能,我們比較幾種抽取方法,希望可以實驗室常備簡單試劑,來有效的萃取沙門氏菌的genomic DNA。
材料與方法
將細菌genomic DNA 萃取出來後,以primer 16S-F 和16S-R做PCR,再以電泳確認所夾取片段大小。經使用yT&A vector 選殖白色菌落培養,並且抽取plasmid,然後以BamHI、EcoRI 兩個限制酶確認後,定序並和資料庫序列進行比對,來鑑定菌種。
結 論
利用alkaline-SDS lysis抽取質體的方法,在加入solution III 離心後,保留pellet,繼續以phenol處理,兩種方法的結合,可以有效萃取出沙門氏菌的genomic DNA,並可以16S rDNA-PCR並定序來鑑定沙氏桿菌[圖一]。
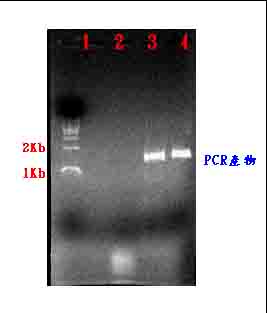
圖一、16S rDNA-PCR凝膠電泳結果。
菌種–抽取genomic DNA方式
1:E.coli(JM109) - alkaline-SDS
2:S.derby - alkaline-SDS
3:E.coli(JM109) - alkaline-SDS及phenol
4:S.derby – alkaline-SDS 及phenol
參考文獻
[1] Cook et al., J Clin Microbiol, 2003, 41:1010.
[2] Knox et al., J Clin Microbiol, 1988, 36:3492.
MC-TB 2010 Vol 2, BTHM2010-c9
萃取弧菌屬細菌染色體DNA用於16S rDNA PCR
陳意心、張猷忠*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:張猷忠[ d80106@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-4-9
中文摘要
食物中毒常為感染腸炎弧菌所引起,因此對於食物中毒的檢體快速鑑定是非常重要的。一般傳統細菌鑑定皆是以生化試驗為主,但這有時會誤判且耗時。由於現今分子生物技術的進步,以PCR 放大細菌內16S rDNA,藉由BLAST 的比對,可快速鑑定出菌種且正確性較高。但不同細菌細胞壁成分常會影響genomic DNA的抽取純度及其後PCR的成功。實驗發現以protease K和phenol處理alkaline-SDS lysis方法所得到的genomic DNA,可成功作為PCR模板。
緒 論
半數以上台灣常見的食物中毒為感染腸炎弧菌所引起[1]。海產食物為重要食物來源之一,容易因為清洗不乾淨且未經烘煮便食用而造成感染[2]。由於造成食物中毒細菌種類不同,可能影響治療,因此檢體的快速鑑定很重要。一般傳統鑑定細菌都是以生化試驗的方式去分類細菌,然而這種方式需要繁雜的培養和生化測試流程,且結果又並非完全正確[3]。現今已知道在各種細菌的16S ribosomal RNA gene 上有保守度較高的片段,也有變異度較高的區域。利用PCR 技術,可藉由universal primer 針對這些保守度較高的片段黏合並放大gene,定序後作BLAST 的比對,藉由這些變異度較高的區域就可以比對出這段16S ribosomal RNA gene 是屬於哪個菌種[4]。
材料與方法
將弧菌培養後,抽取genomic DNA,經由PCR放大16S rDNA 後,跑膠並萃取PCR產物。接著,將16S rDNA 片段與T/A clonging vector 做liagation,再將此vector與已製備好的勝任細胞做transfomation,培養於藍白篩選的培養基內,培養後挑取白色菌落,並利用限制酶切解,再次確認有無成功insert,確認無誤後,將菌送至定序,並將結果與NCBI資料庫進行比對,以便鑑定菌種。
結 論
將萃取的genomic DNA經由PCR放大16S rDNA 片段後,跑膠分析結果與預期1.4 kb 相符合。接著選殖PCR產物,將有insert 的菌送定序後,其結果與NCBI的資料庫進行BLAST比對,結果與Vibrio parahemolyticus有98%相似度。此實驗開始,曾嘗試利用各種方式萃取genomic DNA,但都無法有效萃取出來。主要在於萃取genomic DNA 會因各菌種細胞壁組成的不同而有所影響,革蘭氏陽性菌又比陰性菌更難萃取,最後藉由改良alkaline SDS method,終將genomic DNA萃取出來。
此外本實驗的另一目的是想找出一個穩定萃取genomic DNA 的方法,以便於不同人操作下也能順利萃取genomic DNA。
參考文獻
[1] Boyd et al, BMC Microbiol. 2008, 30:110.
[2] Lynch et al, Infect Immun. 2005, 73:1275.
[3] Christensen et al, J Clin Microbiol. 2003, 41:3790-800.
[4] Bosshard et al, J Clin Microbiol. 2006, 44:1359.
MC-TB 2010 Vol 2, BTHM2010-c10
建立快速篩選可抑制人類免疫缺陷病毒核酸嵌入酶活性的藥物平臺
廖家男、張猷忠*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:張猷忠[ d80106@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-4-9
中文摘要
本實驗利用酵素免疫分析的方法,建立能快速篩選可抑制愛滋病病毒核酸嵌入酶活性藥物的平台。首先由重組質體表現maltose-binding protein(MBP)核酸嵌入酶融合蛋白,目前已成功表現MBP 核酸嵌入酶融合蛋白,並確認該蛋白於大腸桿菌表現時為可溶性。
緒 論
人類免疫缺陷病毒(Human Immunodeficiency Virus, HIV)是一種感染人類免疫系統細胞的慢病毒 (Lentivirus),屬反轉錄病毒(retroviruses)的一種。其特色是利用反轉錄酶(reverse transcriptase)將病毒遺傳物質(ssRNA)反轉錄成去氧核糖核酸(dsDNA),再利用病毒核酸嵌入酵素將病毒dsDNA 隨機嵌入宿主染色體內。抗病毒藥物的研發常以病毒酵素作為標的,如愛滋病毒的反轉錄酶及蛋白酶(protease) 已有不少藥物研發並上市,唯愛滋病毒突變快,故仍需積極開發新的抗病毒藥物。
目前愛滋病毒嵌入酶較少開發藥物,若能開發出抗病毒藥物,雞尾酒療法就可針對三種酵素同時施予藥物治療,或能提高療效,降低抗藥病毒的產生機率。本實驗利用酵素免疫分析的方法(EIA-like method),建立能快速篩選可抑制愛滋病病毒核酸嵌入酶活性藥物的平台[3]。將可用於快速檢測哪些分子對愛滋病病毒核酸嵌入酶有抑制作用,也可與製藥廠或化學公司合作,將其合成的未知化學分子做篩選,加速尋找出有潛力作為日後治療愛滋病的藥物。
材料與方法
本實驗所用材料為pMAL-c2x 質體(具有MBP 融合蛋白質)、pMAL-c2x-IN 質體(具有MBP-IN 融合蛋白質可產生核酸嵌入酶蛋白)及pUC19 質體(嵌入酶蛋白活性測試時用以當作target DNA)。實驗簡略流程如下:先製備核酸嵌入酶蛋白,再以SDS-PAGE 辨認重組蛋白,接著將gel 轉漬至PVDF membrane,西方墨點法辨認。然後萃取pUC19做為target DNA,並以HIV的LTR序列設計出一條經biotin-labeled 的雙股oligo 做為嵌入序列,再以EIA 方式進行嵌入酶蛋白活性測試。
結 論
在誘導表現核酸嵌入酶後,跑SDS-PAGE 電泳確認是有表現的;然後再利用西方墨點法,確認所表現的產物確實為HIV 核酸嵌入酶,之後進行大量表現並分析核酸嵌入酶在大腸桿菌表現,確認該融合蛋白質為可溶性。隨後將進行嵌入酶活性分析。
參考文獻
[1] Excler JL and Indian J, Med Res, 2005, 121:568..
[2] Hu, SL, Curr Drug Targets Infect Disord , 2005, 5:193.
[3] Chang et al, J Virol Methods, 1996, 59:135.
MC-TB 2010 Vol 2, BTHM2010-c11
諾羅病毒#431株的VP1基因選殖
戴薏玲、張猷忠*
銘傳大學健康科技學院生物科技學系(台灣 桃園)
通訊作者:張猷忠[ d80106@mail.mcu.edu.tw ]
收稿:2010-3-10 接受:2010-4-9
中文摘要
諾羅病毒(Norovirus)是造成急性腸胃炎之重要病原之一,為正股RNA 病毒,其Genogroup 可分為五群:GI、GII、GIII、GIV、GV,其中GI 、GII 和GIV 可感染人類,GIII 則感染豬。其主要與感染生物的部位是capsid 的結構蛋白VP1。因此本實驗將從疑似遭諾羅病毒感染病人的檢體中,選殖出諾羅病毒的VP1,再以藍白篩檢定及限制酶切割圖譜的方法確定選殖出之基因,之後將VP1 進行表現,並當作抗原,可用來篩檢血清抗體,了解流行病學,有益日後預防諾羅病毒。
緒 論
諾羅病毒(Norovirus)屬於杯狀病毒科[2],為positive-sense single-stranded RNA,其基因全長約7.2~7.5kbp,有3個ORF (open reading frame):ORF1 是nonstructural protein 的部分,如RdRp (RNA-dependent RNA polymerase);ORF2 是major capsid protrin (VP1);ORF3 是minor capsid protein (VP2)[1]。近年來發現norovirus 為非細菌性急性腸胃炎的主要感染源,具高度的感染性,只需約百個病毒顆粒即可造成感染[3],所有年齡層皆有可能遭到感染,以年輕人為主,在任何季節都可能
引起感染,但主要在冬季。傳染途徑主要以手、糞、口為主,預防措施為勤洗手,尤其進食前及排便後,一定要切實的洗淨雙手,避免直接接觸患者的嘔吐物及排泄物。為求確實預防,所以希望能先了解該病毒的流行概況。本實驗將選殖病毒的VP1 基因當成抗原,利用血清抗體篩檢,來了解病毒的流行概況。
材料與方法
長庚兒童醫院提供疑似遭諾羅病毒感染病人的檢體#431。以PCR 選殖含有RdRp+VP1 序列的DNA到質體pUC19,接著設計VP1 primer 後,以PCR選殖VP1到pMAL-c2x,確認後,表現蛋白當成抗原。
結 論
目前已將諾羅病毒的VP1 增幅出來,但仍在進行選殖轉型。
參考文獻
[1] Rohayem et al, J. Virol, 2005, 79:4977.
[2] Parker et al, J. Virol, 2005, 79:7402.
[3] Christiane et al, J. Virol, 2005, 80:5104.
|